Androgen Insensitivity Syndrome
Back to TopSummary
Sex determination in mammals is dependent on sex chromosome composition, with XX animals being female and XY animals being male. In the absence of the Y-chromosome male determining gene SRY, no androgen is produced in the developing gonad, and the embryo develops as a female. In the presence of the Y-chromosome male determining gene SRY, the developing gonad produces androgen, and the embryo develops as a male. Male development requires a functional androgen receptor on the surface of embryonic cells. Mutations that affect the function of the Androgen Receptor (AR) impair the ability of embryonic cells to initiate male development, transforming chromosomal males (XY) into sterile phenotypic females.
Sterile phenotypic females are obviously unsuitable for breeding, but are otherwise healthy.
Date of Last Update: 04/02/2016
Results
Understanding the Results
Results of the genetic test for Androgen Insensitivity Syndrome are presented as shown below.
| Androgen Insensitivity Syndrome | ||
|---|---|---|
| N/N | Clear | This horse tested negative for AR-M1V. |
| N/Ar | Carrier | Both the normal and mutant alleles are present. This horse is positive for the AR-M1V mutation but will not develop symptoms. |
| Ar/Ar | Affected | This horse carries two copies of the AR-M1V mutation. Genetic males (XY) will be transformed into sterile phenotypic females. Genetic females (XX) will not be affected. |
Disease Name and Genes
Androgen Insensitivity Syndrome is the cause of a Disorder of Sexual Development (DSD) in 64, XY, SRY-positive phenotypic females. Androgen Insensitivity Syndrome is associated with an A to G substitution in the Androgen Receptor (AR) gene that causes a Methionine (M) to Valine (V) substitution in the first codon of the gene (AR-M1V). The substitution of the GTG (V) codon for the ATG (M) codon eliminates the normal start codon for this gene, resulting in a lower level of expression of the Androgen Receptor protein.
Inheritance
Androgen Insensitivity Syndrome is inherited as a recessive disorder. It is caused by a recessive variant of the androgen receptor gene (AR-M1V). The recessive allele is abbreviated here as Ar, with the dominant wild-type allele abbreviated as N.
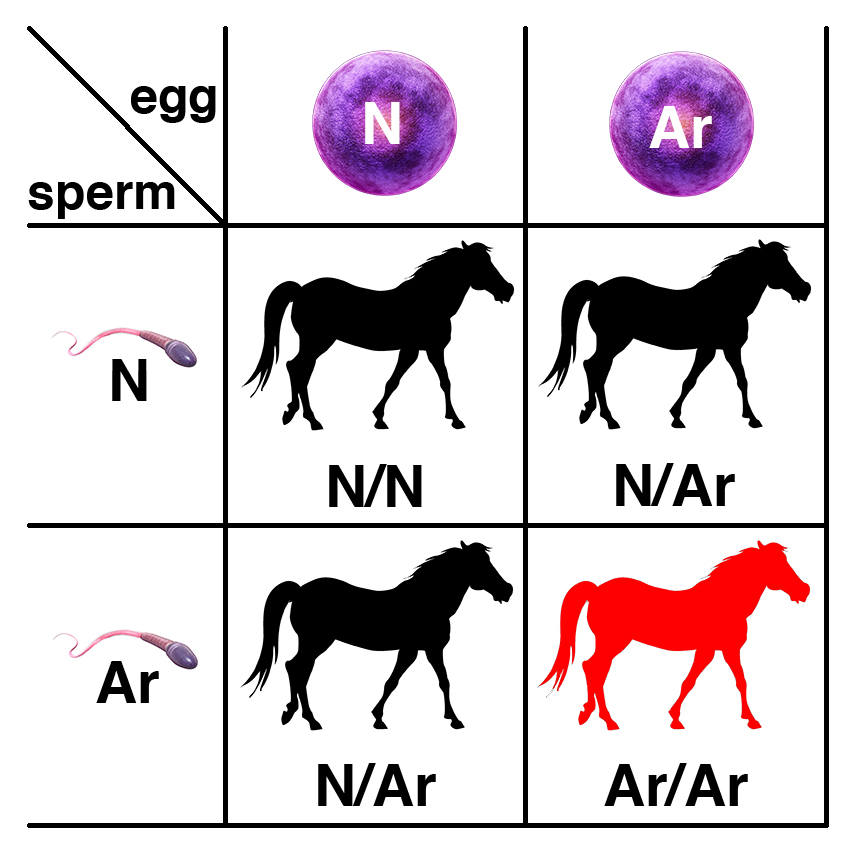
Carriers of the recessive allele (N/Ar) have no symptoms of the disease. If two carriers are bred, each foal has a 25% chance of having two copies of the normal allele (N/N), a 50% chance of being a carrier (N/Ar), and a 25% chance of potentially being affected (Ar/Ar). Only males are affected: although XY, they will appear as 64, XY, SRY-positive phenotypic females that are sterile.
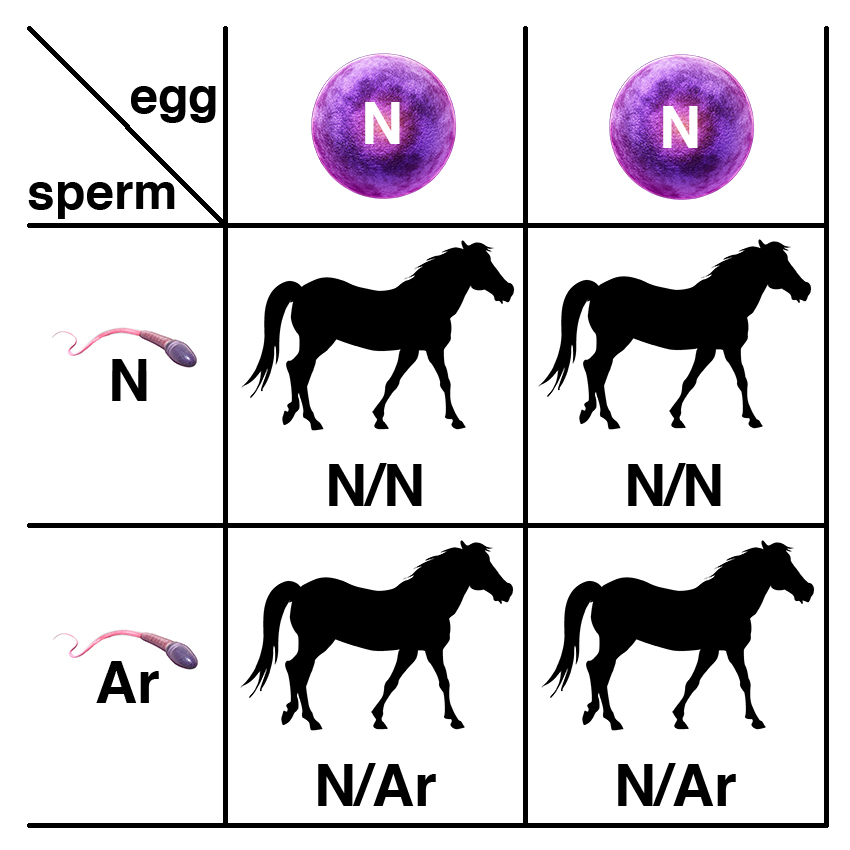
If a carrier of the recessive allele (N/Ar) is bred to a normal horse (N/N), each foal has a 50% chance of having two copies of the normal allele (N/N) and a 50% chance of being a carrier (N/Ar).
Citations
Cerebellar Abiotrophy (CA)
Back to TopSummary
Cerebellar Abiotrophy (CA) is an inherited neurodegenerative disease that causes neurons known as Purkinje cells, located in the cerebellum, to die off. Loss of these neurons affects balance and coordination. Symptoms typically appear between six weeks and four months of age. The symptoms include ataxia (lack of muscle control during voluntary movement, most notably affecting gait), head tremors, and lack of balance. Affected horses are often unable to rise from a reclining position. While the disease is not fatal, most owners elect to euthanize affected horses as their lack of coordination makes handling them dangerous for both the horses and handlers. There is no effective treatment.
Cerebellar Abiotrophy in known primarily in Arabian horses, where carrier rates are estimated to approach 20%.
Date of Last Update: 08/02/2016
Results
Understanding the Results
Results of the genetic test for CA are presented as shown below.
| Cerebellar Abiotrophy (CA) | ||
|---|---|---|
| N/N | Clear | This horse tested negative for CA. |
| N/CA | Carrier | Both the normal and mutant alleles are present. This horse is positive for the CA mutation but will not develop symptoms. |
| CA/CA | Affected | This horse carries two copies of the CA mutation and will develop the disease. |
Disease Name and Genes
Cerebellar Abiotrophy (CA) is caused by a C to T substitution in the upstream regulatory sequence of MUTYH, located approximately 1200 bp upstream of the start codon in a GATA-2 binding site. The substitution results in reduced levels of MUTYH mRNA.
Inheritance
Cerebellar Abiotrophy (CA) is caused by a recessive variant of the MUTYH gene. The recessive allele is commonly abbreviated as CA, with the dominant wild-type allele abbreviated as N.

Carriers of the recessive allele (N/CA) have no symptoms of the disease. If two carriers are bred, each foal has a 25% chance of having two copies of the normal allele (N/N), a 50% chance of being a carrier (N/CA), and a 25% chance of being affected (CA/CA).

If a carrier of the recessive allele (N/CA) is bred to a normal horse (N/N), each foal has a 50% chance of having two copies of the normal allele (N/N) and a 50% chance of being a carrier (N/CA).
Citations
Coat color
Back to TopResults
Citations
Coat color
Back to TopResults
Citations
Coat color
Back to TopResults
Citations
Coat color
Back to TopResults
Citations
Coat color
Back to TopResults
Citations
Coat color
Back to TopResults
Citations
Coat color
Back to TopResults
Citations
Coat color
Back to TopResults
Citations
Coat color
Back to TopResults
Citations
Coat color
Back to TopResults
Citations
Coat color
Back to TopResults
Citations
Congenital Hepatic Fibrosis
Back to TopSummary
Franches-Montagnes horses with Congenital Hepatic Fibrosis show clinical signs of severe liver injury as foals within the first year of life. They are small for their age and show a potbelly due to an increased liver size. The liver shows diffuse fibrosis. There is no effective treatment, and affected foals are typically euthanized.
All affected Franches-Montagnes horses trace their ancestry to the stallion Elu, born in 1964, and have him as both a maternal and paternal ancestor.
Date of Last Update: 03/29/2016
Results
Disease Name and Genes
Congenital Hepatic Fibrosis in Franches-Montagnes horses is associated with variants in the PKHD1 gene, although there is no single nonsynonymous substitution that shows a perfect association (some horses homozygous for each specific variant identified do not develop Congenital Hepatic Fibrosis).
Inheritance
Congenital Hepatic Fibrosis in Franches-Montagnes horses caused by mutations in the PKHD1 gene shows a recessive mode of inheritance.
The recessive allele is abbreviated here as CHF, with the dominant wild-type allele abbreviated as N.

Carriers of the recessive allele (N/CHF) have no symptoms of the disease. If two carriers are bred, each foal has a 25% chance of having two copies of the normal allele (N/N), a 50% chance of being a carrier (N/CHF), and a 25% chance of being affected (CHF/CHF).

If a carrier of the recessive allele (N/CHF) is bred to a normal horse (N/N), each foal has a 50% chance of having two copies of the normal allele (N/N) and a 50% chance of being a carrier (N/CHF).
Citations
Congenital Stationary Night Blindness (CSNB)
Back to TopSummary
Congenital Stationary Night Blindness (CSNB) is an inherited condition characterized by impaired vision in dark conditions. It is present at birth and is nonprogressive. There are no ophthalmic abnormalities on clinical examination of affected horses; diagnosis is confirmed by electroretinography (ERG). The condition is associated with homozygosity for the Leopard Complex spotting allele (LP) that is common in Appaloosas.
Date of Last Update: 08/02/2016
Results
Understanding the Results
Results of the genetic test for CSNB are presented as shown below.
| Congenital stationary night blindness (CSNB) | ||
|---|---|---|
| lp/lp | Clear | This horse tested negative for LP. |
| LP/lp | Carrier | Both the normal and mutant alleles are present. This horse is positive for the LP mutation but will not develop symptoms. |
| LP/LP | Affected | This horse carries two copies of the LP mutation and will develop CSNB. |
Disease Name and Genes
Congenital Stationary Night Blindness (CSNB) is caused by a 1378 bp insertion in the Transient Receptor Potential Cation Channel, Subfamily M, Member 1 (TRPM1) gene. The insertion is a long terminal repeat (LTR) of an endogenous retrovirus that disrupts TRPM1 transcription by premature polyadenylation. While CSNB is fully recessive, this TRPM1 allele produces a dominant white-spotting phenotype, Leopard Complex spotting (LP).
Inheritance
Congenital stationary night blindness (CSNB) is caused by a recessive variant of the TRPM1 gene that has a dominant effect on coat color. The variant allele that is recessive with respect to CSNB is dominant with respect to coat color, and is commonly abbreviated as LP, with the wild-type allele abbreviated as lp.
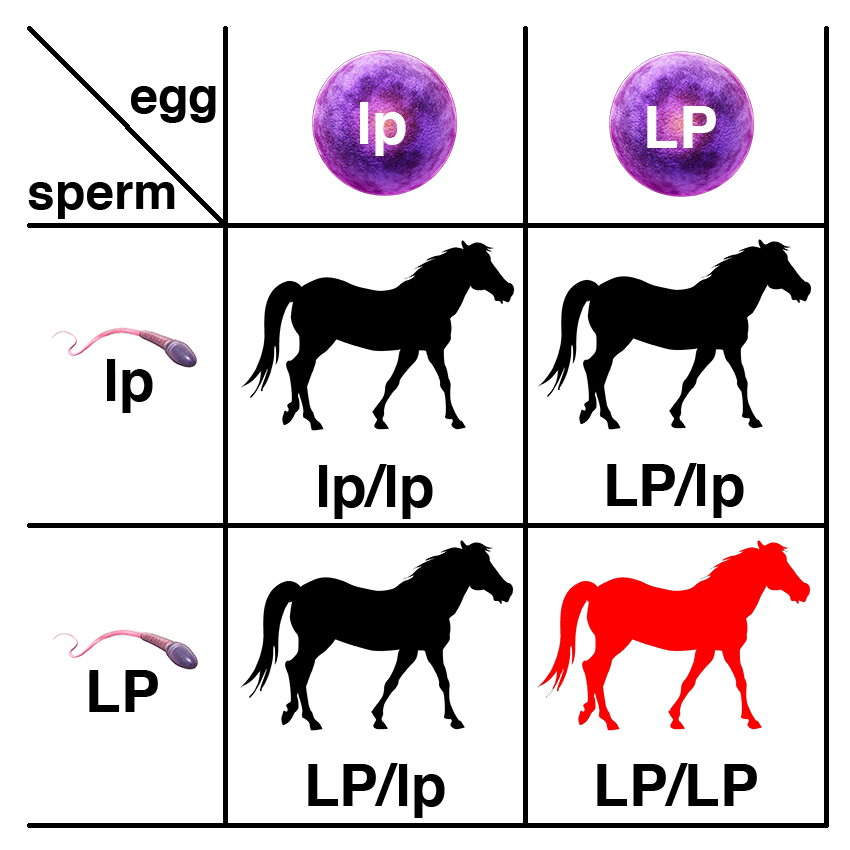
Carriers of the recessive allele (LP/lp) have no symptoms of the disease. If two carriers are bred, each foal has a 25% chance of having two copies of the normal allele (lp/lp), a 50% chance of being a carrier (LP/lp), and a 25% chance of being affected (LP/LP).
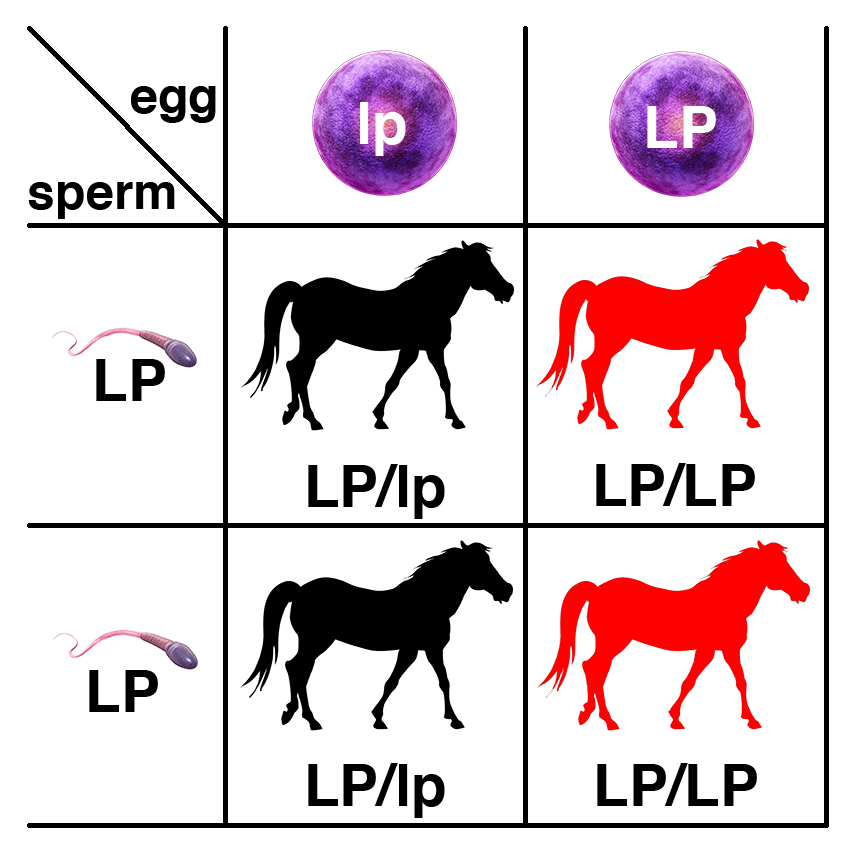
If an affected horses (LP/LP) is bred to a carrier (LP/lp), each foal has a 50% chance of being a carrier (LP/lp), and a 50% chance of being affected (LP/LP).

If two affected horses (LP/LP) are bred, every foal will be affected (LP/LP).

If a carrier of the recessive allele (LP/lp) is bred to a normal horse (lp/lp), each foal has a 50% chance of having two copies of the normal allele (lp/lp) and a 50% chance of being a carrier (LP/lp).
Citations
Dwarfism
Back to TopResults
Citations
Foal Immunodeficiency Syndrome (FIS)
Back to TopSummary
Foal Immunodeficiency Syndrome (FIS) is an inherited disorder of the immune system that manifests as B-lymphocyte immunodeficiency and progressive anemia. Affected foals appear to be normal at birth, but develop infections within a few weeks. The infections are resistant to treatment, and foals die or are euthanized by three months of age.
The condition is found in two rare related British pony breeds, the Fell and the Dales. A significant percentage (10%) of the Fell ponies born each year die from FIS.
Date of Last Update: 08/02/2016
Results
Understanding the Results
Results of the genetic test for FIS are presented as shown below.
| Foal Immunodeficiency Syndrome (FIS) | ||
|---|---|---|
| N/N | Clear | This horse tested negative for FIS. |
| N/FIS | Carrier | Both the normal and mutant alleles are present. This horse is positive for the FIS mutation but will not develop symptoms. |
| FIS/FIS | Affected | This horse carries two copies of the FIS mutation and will die of the disease. |
Disease Name and Genes
Foal Immunodeficiency Syndrome (FIS) is associated with a Proline (P) to Leucine (L) substitution at amino acid position 446 of the sodium/myo-inositol cotransporter gene (SLC5A3-P446L).
Inheritance
Foal Immunodeficiency Syndrome (FIS) is caused by a recessive variant of the SLC5A3 gene (SLC5A3-P446L). The recessive allele is commonly abbreviated as FIS, with the dominant wild-type allele abbreviated as N.
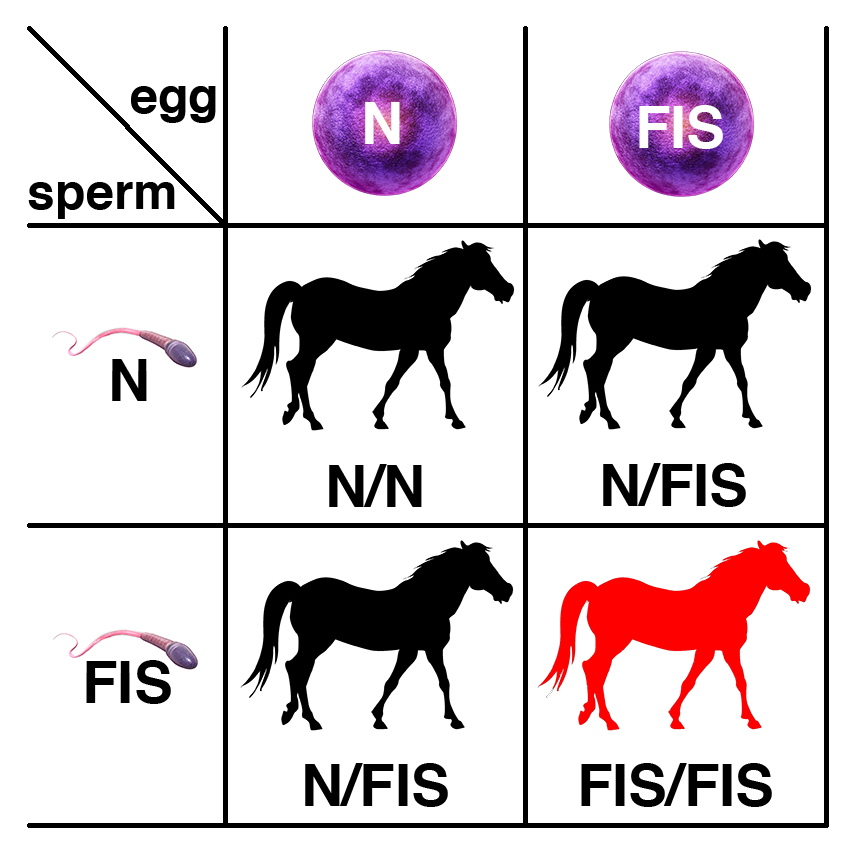
Carriers of the recessive allele (N/FIS) have no symptoms of the disease. If two carriers are bred, each foal has a 25% chance of having two copies of the normal allele (N/N), a 50% chance of being a carrier (N/FIS), and a 25% chance of being affected (FIS/FIS).

If a carrier of the recessive allele (N/FIS) is bred to a normal horse (N/N), each foal has a 50% chance of having two copies of the normal allele (N/N) and a 50% chance of being a carrier (N/FIS).
Citations
Gait
Back to TopResults
Citations
Glycogen Branching Enzyme Deficiency (GBED)
Back to TopSummary
Glycogen branching enzyme deficiency (GBED) is an inherited disease that leads to stillbirths and spontaneous abortions, and cardiac or respiratory failure, seizures, muscle weakness, and sudden death or euthanasia in foals under 18 weeks old. GBED results from a variant of the gene encoding the glycogen branching enzyme, GBE1. The variant form of this enzyme is unable to form α1,6 glycogen branches, leading to an inability to store functional glycogen in muscle and liver. Glycogen is used as energy storage, and is converted to glucose to provide energy to the body between meals, particularly the heart, skeletal muscles, and brain. Lack of energy to these tissues leads to heart, lung, and muscle weakness and failure typical of the disease. Many embryos affected with the disease will die in utero. Most foals affected with the disease will die within eight weeks. There is no treatment for the disease.
About 8.3% of Quarter Horses and 7.1% of Paint Horses have been found to be heterozygotes (carriers) of GBED, and all are related. At least 3% of spontaneous abortions in Quarter Horses are caused by GBED. No homozygous horses have lived more than 18 weeks. The American Quarter Horse Association requires genetic testing for GBED and four other genetic conditions (MH, PSSM1, HYPP, and HERDA).
Date of Last Update: 08/02/2016
Results
Understanding the Results
Results of the genetic test for GBED are presented as shown below.
| Glycogen branching enzyme deficiency (GBED) | ||
|---|---|---|
| N/N | Clear | This horse tested negative for GBE1-T34X and does not carry the GBED mutation identified in Quarter Horses. |
| N/Gb | Carrier | Both the normal and mutant alleles are present. This horse is positive for the GBE1-T34X mutation but will not develop symptoms. |
| Gb/Gb | Affected | This horse carries two copies of the GBE1-T34X mutation and will die of the disease. |
Disease Name and Genes
Glycogen branching enzyme deficiency, a glycogen storage disease also termed GSD IV, is caused by a C to A substitution at base 102 of the gene for glycogen branching enzyme GBE1. The change creates a termination codon in the codon 34 of the 699 amino acid GBE1 protein (GBE1-T34X). The variant is fully recessive.
Inheritance
GBED is caused by a recessive variant of the GBE1 gene (GBE1-T34X). The recessive allele is commonly abbreviated as Gb, with the dominant wild-type allele abbreviated as N.

Carriers of the recessive allele (N/Gb) have no symptoms of the disease. If two carriers are bred, each foal has a 25% chance of having two copies of the normal allele (N/N), a 50% chance of being a carrier (N/Gb), and a 25% chance of being affected (Gb/Gb).

If a carrier of the recessive allele (N/Gb) is bred to a normal horse (N/N), each foal has a 50% chance of having two copies of the normal allele (N/N) and a 50% chance of being a carrier (N/Gb).
Resources
Citations
Hereditary Equine Regional Dermal Asthenia (HERDA)
Back to TopSummary
Hereditary equine regional dermal asthenia (HERDA), or hyperelastosis cutis (HC) is an inherited condition causing skin lesions and ulcerations, poor healing and scarring of wounds, and skin sloughing along the back of affected horses beginning around 1.5-2 years of age and progressively worsening over time. This disease has been found in Quarter Horses and related breeds. Most affected horses are euthanized as they cannot be ridden and are not suitable for breeding. There is no effective treatment. The condition resembles Ehlers-Danlos syndrome in humans, which has multiple genetic causes. The skin defects resemble those seen in the Warmblood Fragile Foal Syndrome (WFFS), which has an earlier onset of symptoms. There are also reports of Ehlers-Danlos-like syndromes of unknown origin in a number of breeds.
The carrier frequency is about 3.5% in Quarter Horses, with an incidence of over 25% in Cutting horses. It is especially prevalent among horses descended from the Quarter Horse sire, Poco Bueno. The American Quarter Horse Association requires genetic testing for HERDA and four other genetic conditions (GBED, PSSM1, MH, and HYPP).
Date of Last Update: 08/02/2016
Results
Understanding the Results
Results of the genetic test for HERDA are presented as shown below.
| Hereditary equine regional dermal asthenia (HERDA) | ||
|---|---|---|
| N/N | Clear | This horse tested negative for HERDA. |
| N/Hrd | Carrier | Both the normal and mutant alleles are present. This horse is positive for the PPIB-G39R mutation but will not develop symptoms of HERDA. |
| Hrd/Hrd | Affected | This horse carries two copies of the PPIB-G39R mutation and will develop HERDA. |
Disease Name and Genes
Hereditary equine regional dermal asthenia (HERDA) is caused by a G to A substitution at base 115 of the gene for peptidyl-prolyl isomerase B (PPIB), causing a glycine to arginine substitution at amino acid position 39 in the PPIB protein (PPIB-G39R).
Inheritance
HERDA is caused by a recessive variant of the PPIB gene (PPIB-G39R). The recessive allele is commonly abbreviated as Hrd, with the dominant wild-type allele abbreviated as N.

Carriers of the recessive allele (N/Hrd) have no symptoms of the disease. If two carriers are bred, each foal has a 25% chance of having two copies of the normal allele (N/N), a 50% chance of being a carrier (N/Hrd), and a 25% chance of being affected (Hrd/Hrd).

If a carrier of the recessive allele (N/Hrd) is bred to a normal horse (N/N), each foal has a 50% chance of having two copies of the normal allele (N/N) and a 50% chance of being a carrier (N/Hrd).
Resources
Citations
Hoof Wall Separation Disease (HWSD)
Back to TopSummary
Hoof Wall Separation Disease (HWSD) is an inherited condition observed in Connemara ponies. Affected animals show separation and breaking of the dorsal hoof wall along the weight-bearing surface of the hoof during the first year of life. Loss of the structural integrity of the hoof makes affected horses unable to support their weight effectively. Chronic inflammation often leads to laminitis, a debilitating condition in which the bone separates from the hoof wall. This causes extreme pain and disability, often requiring euthanasia. While the condition is not fully penetrant (some affected horses have a less severe form of the disease), there is no effective treatment for horses that are strongly affected.
About 15% of Connemara ponies are carriers. Carriers are unaffected.
Date of Last Update: 04/02/2016
Results
Understanding the Results
Results of the genetic test for HWSD are presented as shown below.
| Hoof Wall Separation Disease (HWSD) | ||
|---|---|---|
| N/N | Clear | This horse tested negative for HWSD. |
| N/HD | Carrier | Both the normal and mutant alleles are present. This horse is positive for the HWSD mutation but will not develop symptoms. |
| HD/HD | Affected | This horse carries two copies of the HWSD mutation and will develop Hoof Wall Separation Disease. |
Disease Name and Genes
Hoof Wall Separation Disease (HWSD) is associated with the insertion of a single C in the fifth exon of SERPINB11. The insertion alters the two amino acids following position 168, then introduces a termination codon that truncates 55% of the predicted protein (SERPINB11 c.504_505insC).
Inheritance
Hoof Wall Separation Disease (HWSD) is caused by a recessive variant of the SERPINB11 gene. The recessive allele is commonly abbreviated as HD, with the dominant wild-type allele abbreviated as N.

Carriers of the recessive allele (N/HD) have no symptoms of the disease. If two carriers are bred, each foal has a 25% chance of having two copies of the normal allele (N/N), a 50% chance of being a carrier (N/HD), and a 25% chance of being affected (HD/HD).

If a carrier of the recessive allele (N/HD) is bred to a normal horse (N/N), each foal has a 50% chance of having two copies of the normal allele (N/N) and a 50% chance of being a carrier (N/HD).
Citations
Hydrocephalus in Friesian Horses
Back to TopSummary
Hydrocephalus in Friesian horses is an inherited developmental disorder that often results in stillbirths of affected foals and obstructed labor (dystocia) in dams, which can be fatal to the dam during birth. Hydrocephalus is a distension of the ventricular system of the brain with cerebrospinal fluid. The increased intracranial pressure inside the skull leads to enlargement of the head. The condition is usually fatal. There is no effective treatment.
About 15% of Friesian horses are carriers of the recessive trait. Hydrocephalus has been seen rarely in other breeds, although it is unclear whether it has the same genetic basis.
Date of Last Update: 08/02/2016
Results
Understanding the Results
Results of the genetic test for Hydrocephalus in Friesian horses are presented as shown below.
| Hydrocephalus in Friesian horses | ||
|---|---|---|
| N/N | Clear | This horse tested negative for Hydrocephalus. |
| N/HCP | Carrier | Both the normal and mutant alleles are present. This horse is positive for the Hydrocephalus mutation but will not develop symptoms. |
| HCP/HCP | Affected | This horse carries two copies of the Hydrocephalus mutation and will die of the disease. |
Disease Name and Genes
Hydrocephalus in Friesian horses is associated with a C to T substitution that changes a Glutamine (Q) codon to a termination codon at amino acid position 475 of the B3GALNT2 gene (B3GALNT2-Q475X).
Inheritance
Hydrocephalus in Friesian horses is caused by a recessive variant of the B3GALNT2 gene (B3GALNT2-Q475X). The recessive allele is commonly abbreviated as HCP, with the dominant wild-type allele abbreviated as N.

Carriers of the recessive allele (N/HCP) have no symptoms of the disease. If two carriers are bred, each foal has a 25% chance of having two copies of the normal allele (N/N), a 50% chance of being a carrier (N/HCP), and a 25% chance of being affected (HCP/HCP).
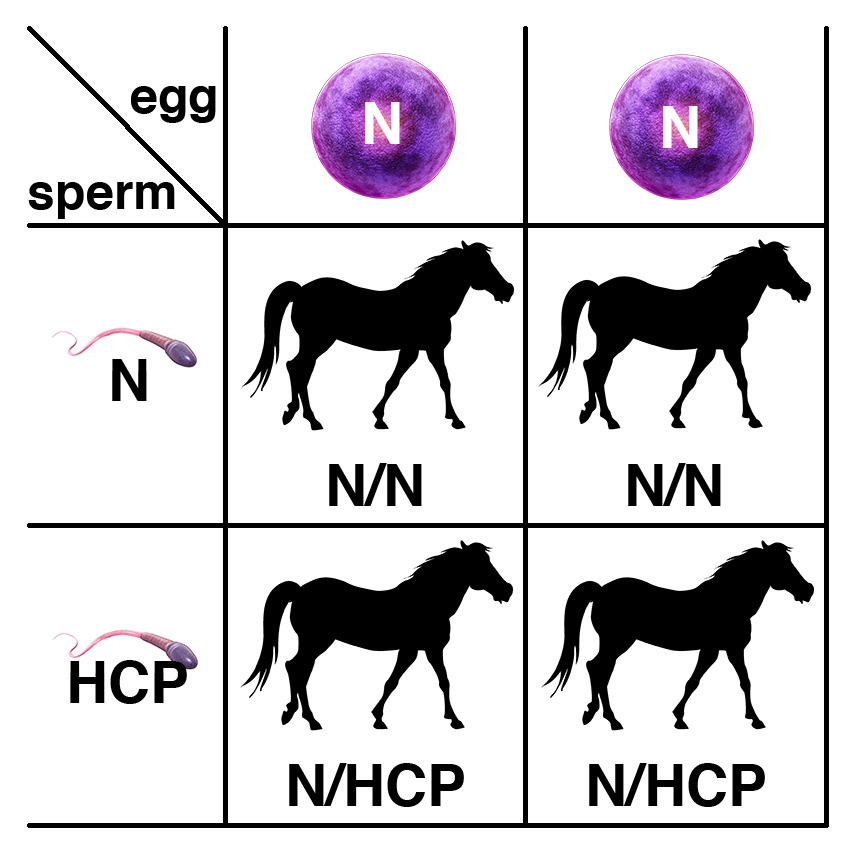
If a carrier of the recessive allele (N/HCP) is bred to a normal horse (N/N), each foal has a 50% chance of having two copies of the normal allele (N/N) and a 50% chance of being a carrier (N/HCP).
Citations
Hyperkalemic Periodic Paralysis (HYPP)
Back to TopSummary
Hyperkalemic periodic paralysis (HYPP) is an inherited disease that causes sporadic episodes of muscle spasms, weakness, or paralysis in affected horses exposed to high potassium levels. Respiratory paralysis or heart failure may lead to death. It has been found in descendants of the American Quarter horse sire, Impressive, including Paints and Appaloosas of this lineage. Episodes may be triggered by stress, diet, lack of exercise, and recovery from anesthesia. HYPP is managed by a low-potassium diet, regular exercise, or drugs (acetazolamide, hydrochlothiazide, or intravenous calcium).
The known genetic cause of HYPP is an autosomal dominant variant of the SCN4A gene that encodes a sodium channel of skeletal muscle. The variant form of this channel fails to regulate potassium levels in the blood leading to uncontrolled muscle contractions. Homozygous horses (with two defective genes) are affected more severely.
Most affected horses are well-muscled halter show horses. Spontaneous muscle contractions in affected horses may cause this desired greater muscle mass. The American Quarter Horse Association requires genetic testing for HYPP and four other genetic conditions (GBED, PSSM1, MH, and HERDA).
Date of Last Update: 08/02/2016
Results
Understanding the Results
Results of the genetic test for HYPP are presented as shown below.
| Hyperkalemic periodic paralysis (HYPP) | ||
|---|---|---|
| n/n | Clear | This horse tested negative for HYPP and does not carry the SCN4A-F1416L mutation. |
| n/H | Affected | Both the normal and mutant alleles are present. This horse is positive for the SCN4A-F1416L mutation and is expected to develop symptoms of HYPP. |
| H/H | Affected | This horse carries two copies of the SCN4A-F1416L mutation and is positive for HYPP. The horse is expected to develop symptoms of HYPP. |
Disease Name and Genes
Hyperkalemic periodic paralysis (HYPP) is caused by a missense mutation in the SCN4A gene that encodes a sodium channel (SCN4A-F1416L). The F1416L substitution occurs in transmembrane domain IV near the cytoplasmic face of the membrane. This mutation causes potassium-induced paralysis associated with myotonia.
Inheritance
HYPP is associated with a dominant variant of the SCN4A gene (SCN4A-F1416L). The dominant allele is commonly abbreviated as H, with the recessive wild-type allele abbreviated as n.

A horse with one copy of the dominant allele (n/H) will have symptoms. If this horse is bred to a normal horse (n/n), each foal has a 50% chance of having one copy of the dominant allele (n/H) and a 50% chance of having two copies of the normal allele (n/n).
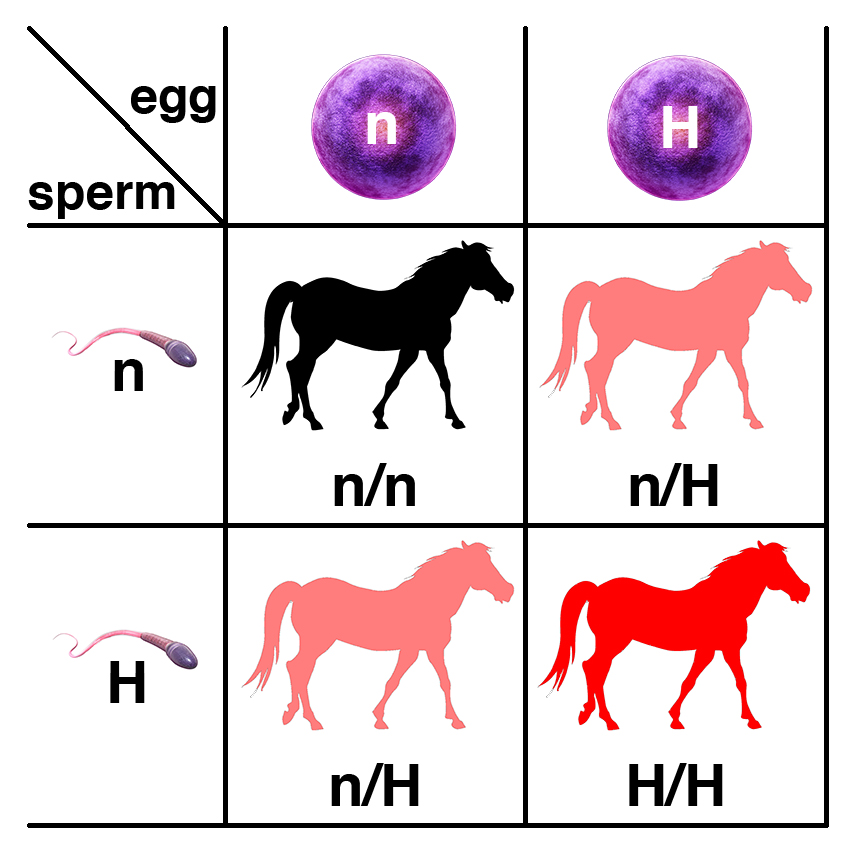
If two horses, each with one copy of the dominant allele (n/H), are bred, each foal has a 25% chance of having two copies of the normal allele (n/n), a 50% chance of having one copy of the dominant allele (n/H), and a 25% chance of having two copies of the semidominant allele (H/H). Because horses with one copy (n/H) or two copies (H/H) of the dominant allele are affected, each foal will have a 75% chance of being affected.
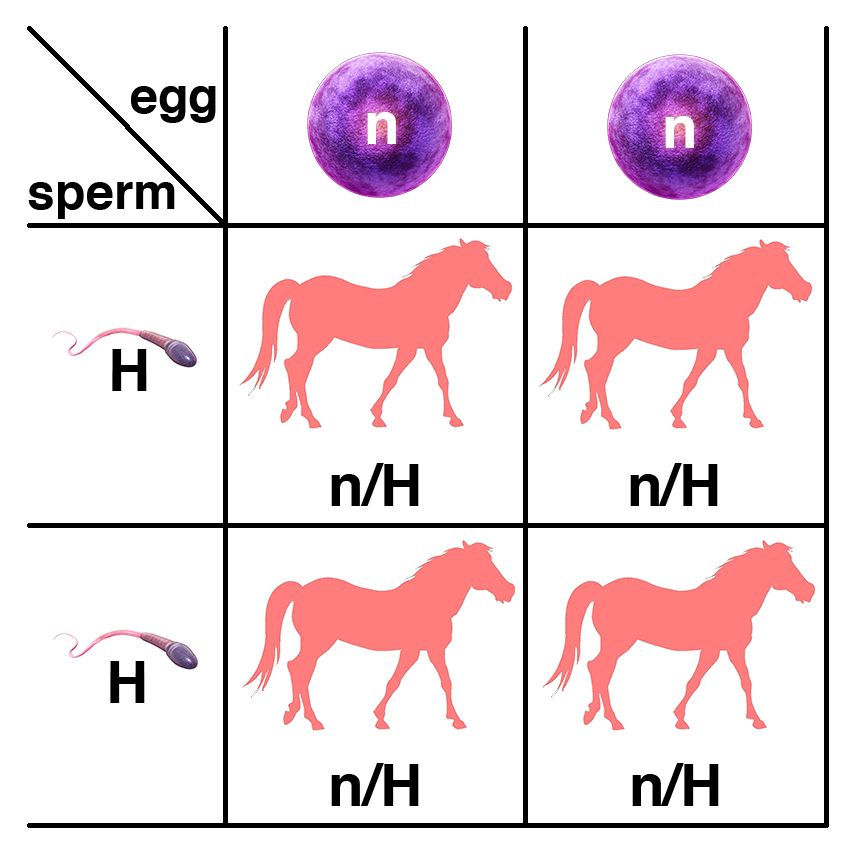
If a horse with two copies of the semidominant allele (H/H) is bred to a normal horse (n/n), all of the foals will have one copy of the semidominant allele (n/H) and will be affected.
Resources
Hyperkalemic periodic paralysis (HYPP) at UC Davis
Citations
Rudolf JA (1992). "Periodic paralysis in quarter horses: a sodium channel mutation disseminated by selective breeding." Nat Genet. 2(2):144-147. PMID: 1338908.Immune-Mediated Myositis
Back to TopSummary
Immune-mediated myositis (IMM) is an autoimmune disease characterized by rapid-onset muscle wasting and the presence of lymphocytes in skeletal muscle. The disease has both environmental and genetic components. The environmental component is recent infection, particularly with Streptococci, or vaccination against influenza, Equine Herpes Virus 4, or strangles (Streptococcus equi). One of the genetic components is MYH1-E321G, a variant of a gene encoding one of the myosin heavy chains.
The MYH1-E321G variant has incomplete penetrance. Not all horses with one or two copies of this variant will develop IMM. There are also horses that appear to have IMM even though they do not have the MYH1-E321G variant; these may be other inflammatory myopathies with a different etiology.
So far, the MYH1-E321G variant has only been identified in Quarter Horses. It was not found in a sample of 175 horses representing 21 breeds not including Quarter Horses.
Date of Last Update: 06/15/2018
Results
Understanding the Results
Results of the genetic test for IMM are presented as shown below.
| Immune-mediated myositis (IMM) | ||
|---|---|---|
| N/N | Clear | This horse tested negative for MYH1-E321G and does not carry the MYH1 mutation identified in Quarter Horses. |
| N/My | Affected | Both the normal and mutant alleles are present. This horse is positive for the MYH1-E321G mutation and may develop symptoms. |
| My/My | Affected | This horse carries two copies of the MYH1-E321G mutation and may develop symptoms. |
What You Can Do
If you have a Quarter Horse that shows rapid muscle loss in the back (epaxial) and rump (gluteal) muscles shortly after an infection or vaccination, you should seek a diagnosis from your veterinarian. If your horse has tested positive for the MYH1-E321G variant, you should share this information with your veterinarian. IMM responds well to treatment with corticosteroids.
Disease Name and Genes
Immune-mediated myositis (IMM) is associated with a missense mutation in the MYH1 gene that encodes one of the myosin heavy chains (MYH1-E321G). The mutation causes a nonconservative amino acid substitution in a highly-conserved position in the myosin head.
Inheritance
IMM is associated with a dominant allele of the MYH1 gene (MYH1-E321G) that has incomplete penetrance. The dominant allele is commonly abbreviated as My, with the recessive wild-type allele abbreviated as N.
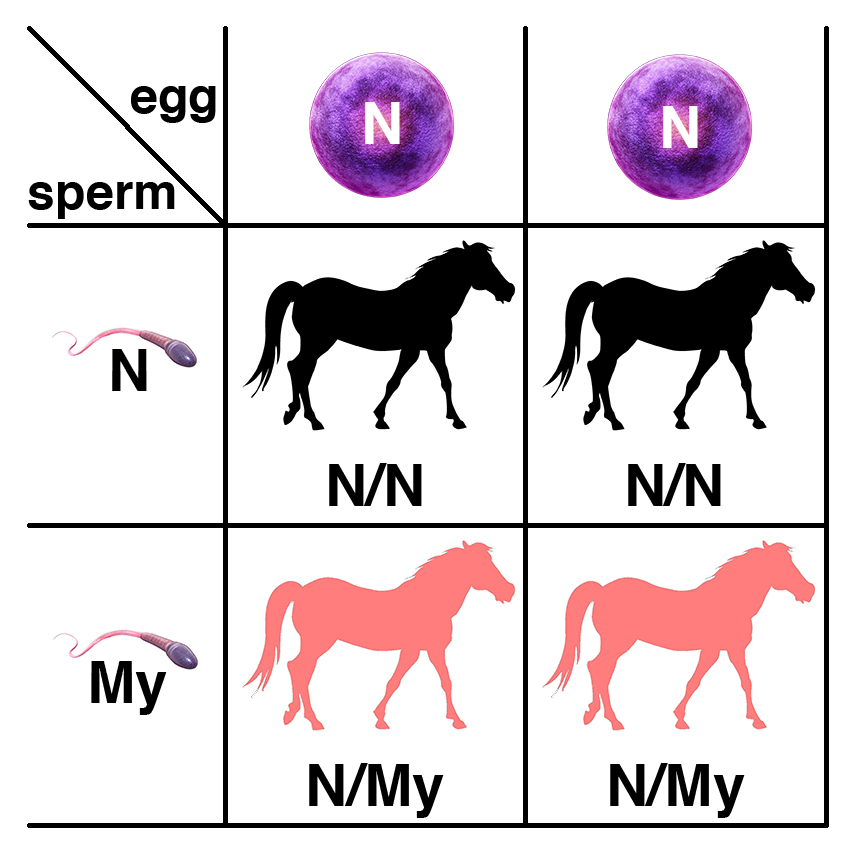
A horse with one copy of the dominant allele (N/My) will potentially have symptoms. If this horse is bred to a normal horse (N/N), each foal has a 50% chance of having one copy of the dominant allele (N/My) and a 50% chance of having two copies of the normal allele (N/N).
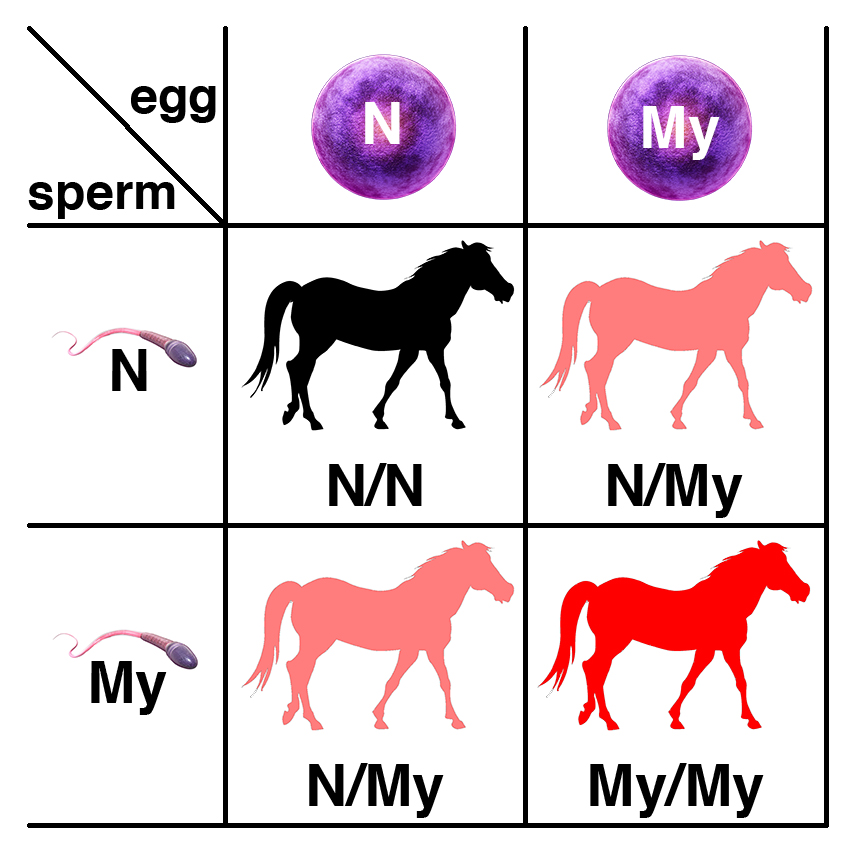
If two horses, each with one copy of the dominant allele (N/My) are bred, each foal has a 25% chance of having two copies of the normal allele (N/N), a 50% chance of having one copy of the dominant allele (N/My), and a 25% chance of having two copies of the dominant allele (My/My). Because horses with one copy (N/My) or two copies (My/My) of the dominant allele have the potential to be affected, each foal will have a 75% chance of potentially being affected.
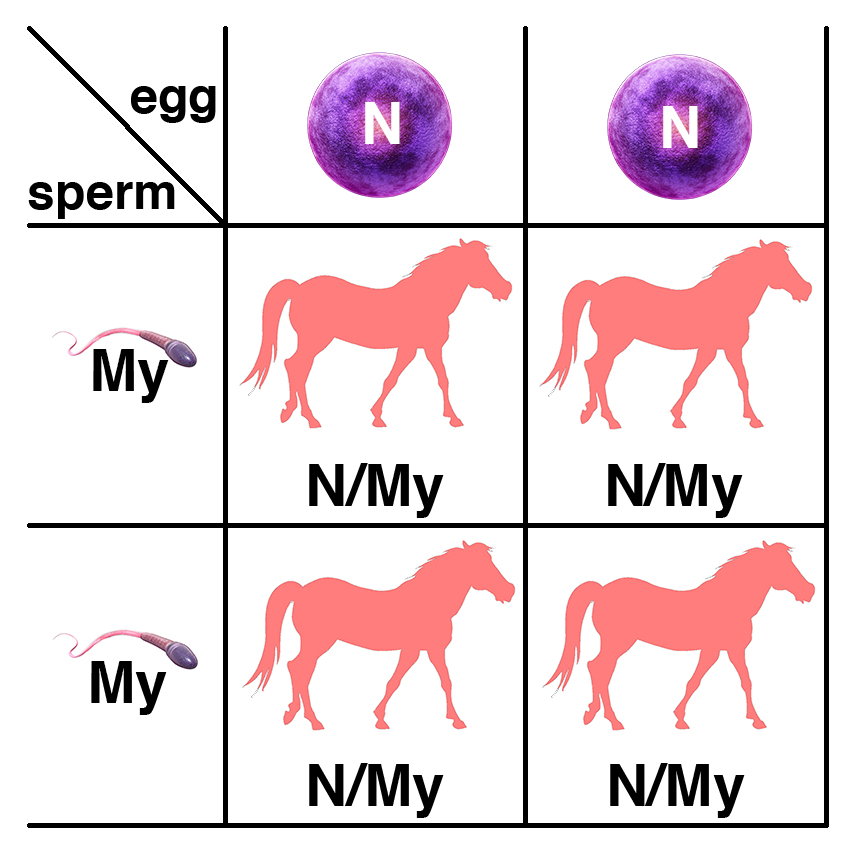
If a horse with two copies of the dominant allele (My/My) is bred to a normal horse (N/N), all of the foals will have one copy of the dominant allele (N/My) and will potentially be affected.
Resources
The Veterinary Genetics Laboratory at UC Davis offers a genetic test for MYH1-E321G.
Citations
Incontinentia Pigmenti
Back to TopSummary
Incontinentia pigmenti (IP) is an inherited skin disorder that causes skin lesions soon after birth that develop into warty areas with hair loss. Wooley hair may grow back in these areas. Affected horses have streaks of light and dark coat color from birth. In addition, there are abnormalities of tooth, hoof, and eye development. Only affected mares have been observed; affected males die before birth.
The condition resembles Ectodermal Dysplasia (ED) in humans. Ectodermal Dysplasia is a collective term for a genetically distinct group of inherited disorders affecting ectodermal structures (skin, nails, and teeth).
Date of Last Update: 08/02/2016
Results
Understanding the Results
Results of the genetic test for Incontinentia Pigmenti (IP) are presented as shown below.
| Incontinentia Pigmenti (IP) | ||
|---|---|---|
| n/n | Clear | This mare tested negative for IP. |
| n/IP | Affected | Both the normal and mutant alleles are present. This mare is positive for the IP mutation and will show symptoms. |
Disease Name and Genes
Incontinentia Pigmenti (IP) is caused by a C to T substitution in the X-chromosomal IKBKG gene that changes an Arginine (R) codon in position 62 to a termination codon (IKBKG-R62X).
Inheritance
Incontinentia Pigmenti (IP) is caused by a dominant variant of the X-chromosomal IKBKG gene (GBE1-T34X). The recessive wild-type allele is abbreviated as n, with the dominant variant abbreviated as IP. Males inheriting the variant (IP/Y) die as embryos.

If a mare with the disorder (n/IP) is crossed to a normal stallion (n/Y), each conception has a 25% chance of being a normal filly (n/n), a 25% chance of being an affected filly (n/IP), a 25% chance of being a normal colt (n/Y), and a 25% chance of being an inviable male embryo (IP/Y).
Citations
Junctional Epidermolysis Bullosa (JEB1 and JEB2)
Back to TopSummary
Junctional Epidermolysis Bullosa (JEB), formerly called epitheliogenesis imperfecta (EI) in horses, is an inherited condition affecting the integrity of the skin. Affected horses have fragile skin that blisters easily. Blistering of the mouth and exungulation (loss of the hoof) are also seen. The condition is evident at birth, and foals are typically euthanized in the first few days of life. There is no effective treatment.
Two distinct genetic forms are known. JEB1, resulting from a mutation in LAMC2, is seen in Belgians, while JEB2, resulting from a mutation in LAMA3, is seen in the American Saddlebred.
Date of Last Update: 04/04/2016
Results
Citations
Lavender Foal Syndrome (LFS)
Back to TopSummary
Lavender Foal Syndrome (LFS) is an inherited condition that causes seizures and the death of foals soon after birth. Symptoms include opisthotonus (a spasm of the muscles causing backward arching of the head, neck and spine) and stiff or paddling leg movements. These foals are unable to stand and nurse and are typically euthanized. There is no effective treatment.
Affected foals have a diluted coat color referred to as "lavender."
Date of Last Update: 08/02/2016
Results
Understanding the Results
Results of the genetic test for LFS are presented as shown below.
| Lavender Foal Syndrome (LFS) | ||
|---|---|---|
| N/N | Clear | This horse tested negative for Lf. |
| N/Lf | Carrier | Both the normal and mutant alleles are present. This horse is positive for the Lf mutation but will not develop symptoms. |
| Lf/Lf | Affected | This horse carries two copies of the Lf mutation and will die of the disease. |
Disease Name and Genes
Lavender Foal Syndrome (LFS) results from a frameshift mutation in the myosin Va (MYO5A) gene. A single-base deletion in exon 30 changes the reading frame and results in a premature stop codon.
Inheritance
Lavender Foal Syndrome (LFS) is caused by a recessive variant of the MYO5A gene. The recessive allele is commonly abbreviated as Lf, with the dominant wild-type allele abbreviated as N.

Carriers of the recessive allele (N/Lf) have no symptoms of the disease. If two carriers are bred, each foal has a 25% chance of having two copies of the normal allele (N/N), a 50% chance of being a carrier (N/Lf), and a 25% chance of being affected (Lf/Lf).

If a carrier of the recessive allele (N/Lf) is bred to a normal horse (N/N), each foal has a 50% chance of having two copies of the normal allele (N/N) and a 50% chance of being a carrier (N/Lf).
Citations
Brooks SA et al. (2010). "Whole-genome SNP association in the horse: identification of a deletion in myosin Va responsible for Lavender Foal Syndrome." PLoS Genet.. 6(4):e1000909. PMID: 20419149.Lethal White Overo (LWO)
Back to TopSummary
Lethal White Overo (LWO), also known as Lethal White Foal Syndrome (LWFS) or Overo Lethal White Syndrome (OLWS), is an inherited condition associated with the Frame Overo patterning gene that affects coat color. Foals affected by Lethal White Overo have aganglionosis (lack of parasympathetic ganglion cells) in the distal small intestine and the large intestine, resulting in megacolon, an abnormal dilation of the colon due to paralysis of the peristaltic movements of the bowel. The condition is similar to the human inherited condition Hirschsprung disease, caused by mutations in the orthologous human gene. Affected foals dies shortly after birth due to intestinal blockage. Their coat color is entirely white. There is no effective treatment.
The perinatal lethality associated with Frame Overo is a recessive characteristic. The coat color phenotype (white spotting) is dominant. The Frame Overo variant is found in Paints, Pintos, Quarter Horses, Miniature Horses, and Thoroughbreds.
Date of Last Update: 08/02/2016
Results
Understanding the Results
Results of the genetic test for Lethal White Overo (LWO) are presented as shown below.
| Lethal White Overo (LWO) | ||
|---|---|---|
| n/n | Clear | This horse tested negative for frame Overo. |
| n/O | Carrier | Both the normal and mutant alleles are present. This horse is positive for the frame Overo mutation but will not develop symptoms. |
| O/O | Affected | This horse carries two copies of the frame Overo mutation and will die of the disease. |
Disease Name and Genes
Lethal White Overo (LWO) or Lethal White Foal Syndrome (LWFS) is caused by a TC to AG dinucleotide mutation of the endothelin-B receptor gene (EDNRB) that causes the replacement of Isoleucine (I) to Lysine (K) at amino acid position 118 of the protein (EDNRB-I118K).
Inheritance
Lethal White Overo (LWO) is a recessive lethal, with affected foals dying shortly after birth. It is caused by a recessive variant of the EDNRB gene (EDNRB-I118K), with the variant commonly abbreviated as O, and the dominant wild-type allele abbreviated as n. The EDNRB-I118K variant produces the frame Overo coat color pattern as a dominant effect.

Carriers of the recessive allele (n/O) have no symptoms of the disease. If two carriers are bred, each foal has a 25% chance of having two copies of the normal allele (n/n), a 50% chance of being a carrier (n/O), and a 25% chance of being affected (O/O).

If a carrier of the recessive allele (n/O) is bred to a normal horse (n/n), each foal has a 50% chance of having two copies of the normal allele (n/n) and a 50% chance of being a carrier (n/O).
Citations
Malignant Hyperthermia (MH)
Back to TopSummary
Malignant Hyperthermia (MH) is an inherited disease that causes severe tying up and possible death in affected horses after exposure to anesthesia. It has been reported in Quarter Horses and related breeds, Thoroughbreds, Arabians, and ponies. Episodes may also be triggered by exercise, stress, or episodes of other myopathies such as PSSM1. Signs include high temperature and profuse sweating, rapid heart rate and breathing, muscle rigidity, and high blood pressure. Fatalities may be prevented by treatment with dantrolene prior to administering anesthesia or application of body-cooling therapy during episodes.
The most common known genetic cause of MH is an autosomal dominant variant of the RYR1 gene that encodes the ryanodine receptor (a calcium release channel) of the sarcoplasmic reticulum of skeletal muscle. The variant form of this channel leads to an uncontrolled release of intracellular calcium from the sarcoplasmic reticulum.
Less than 1% of Quarter Horses are affected, with most being of halter bloodlines. Horses affected with both the GYS1-R309H (PSSM1) and RYR1 (MH) mutation have higher exercise intolerance than those with PSSM1 alone. The American Quarter Horse Association requires genetic testing for MH and four other genetic conditions (GBED, PSSM1, HYPP, and HERDA).
Date of Last Update: 08/02/2016
Results
Understanding the Results
Results of the genetic test for MH are presented as shown below.
| Malignant Hyperthermia (MH) | ||
|---|---|---|
| n/n | Clear | This horse tested negative for MH and does not carry the RYR1-R2454G mutation. |
| n/MH | Affected | Both the normal and mutant alleles are present. This horse is positive for the RYR1-R2454G mutation and is expected to display symptoms of MH. |
| MH/MH | Affected | This horse carries two copies of the RYR1-R2454G mutation and is positive for MH. The horse is expected to develop symptoms of MH. |
Disease Name and Genes
Malignant Hyperthermia (MH) is caused by a missense mutation that replaces an Arginine (R) with a Glycine (G) at amino acid position 2454 in the RYR1 gene that encodes the ryanodine receptor (RYR1-R2454G).
Inheritance
Malignant Hyperthermia (MH) is associated with a dominant variant of the RYR1 gene (RYR1-R2454G). The dominant allele is commonly abbreviated as MH, with the recessive wild-type allele abbreviated as n.

A horse with one copy of the dominant allele (n/MH) will potentially have symptoms. If this horse is bred to a normal horse (n/n), each foal has a 50% chance of having one copy of the semidominant allele (n/MH) and a 50% chance of having two copies of the normal allele (n/n).

If two horses, each with one copy of the dominant allele (n/MH), are bred, each foal has a 25% chance of having two copies of the normal allele (n/n), a 50% chance of having one copy of the semidominant allele (n/MH), and a 25% chance of having two copies of the semidominant allele (MH/MH). Because horses with one copy (n/MH) or two copies (MH/MH) of the dominant allele are affected, each foal will have a 75% chance of being affected.

If a horse with two copies of the semidominant allele (MH/MH) is bred to a normal horse (n/n), all of the foals will have one copy of the semidominant allele (n/MH) and will be affected.
Citations
Multiple Congenital Ocular Anomalies (MCOA)
Back to TopSummary
Multiple Congenital Ocular Anomalies (MCOA) syndrome is an inherited condition affecting horses with Silver coat color dilution. Horses homozygous for the Silver allele have a wide range of ocular defects, including underdevelopment of the iris, congenital glaucoma presenting as megaloglobus (greatly enlarged eyes), iridociliary cysts (uveal cysts, small cysts in the eye), and cataracts. Horses heterozygous for the Silver allele have less severe defects, primarily iridociliary cysts.
Breeds that have been diagnosed with MCOA include the Rocky Mountain Horse (including the two related breeds Kentucky Mountain Saddle Horse and Mountain Pleasure Horse), Icelandics, Shetland Pony, Exmoor Pony, American Miniature, Belgian Draft, and Morgan.
Date of Last Update: 08/02/2016
Results
Understanding the Results
Results of the genetic test for Multiple Congenital Ocular Anomalies (MCOA) syndrome are presented as shown below.
| Multiple Congenital Ocular Anomalies (MCOA) | ||
|---|---|---|
| n/n | Clear | This horse tested negative for MCOA. |
| n/Z | Affected | Both the normal and mutant alleles are present. This horse is positive for the PMEL-R625C mutation and will develop mild symptoms. |
| Z/Z | Affected | This horse carries two copies of the PMEL-R625C mutation and will develop symptoms. |
Disease Name and Genes
Multiple Congenital Ocular Anomalies (MCOA) syndrome is caused by a C to T substitution in the premelanosome protein PMEL that results in a missense mutation that changes an Arginine (R) to Cysteine (C) at codon 625 (PMEL-R625C).
Inheritance
Multiple Congenital Ocular Anomalies (MCOA) syndrome is inherited as a semidominant trait associated with the Silver coat color dilution trait (PMEL-R625C). The semidominant allele is commonly abbreviated as Z, with the recessive wild-type allele abbreviated as n.

A horse with one copy of the semidominant allele (n/Z) will have mild symptoms. If this horse is bred to a normal horse (n/n), each foal has a 50% chance of having one copy of the semidominant allele (n/Z) and a 50% chance of having two copies of the normal allele (n/n).

If two horses, each with one copy of the semidominant allele (n/Z), are bred, each foal has a 25% chance of having two copies of the normal allele (n/n), a 50% chance of having one copy of the semidominant allele (n/Z) and being mildly affected, and a 25% chance of having two copies of the semidominant allele (Z/Z) and being strongly affected.
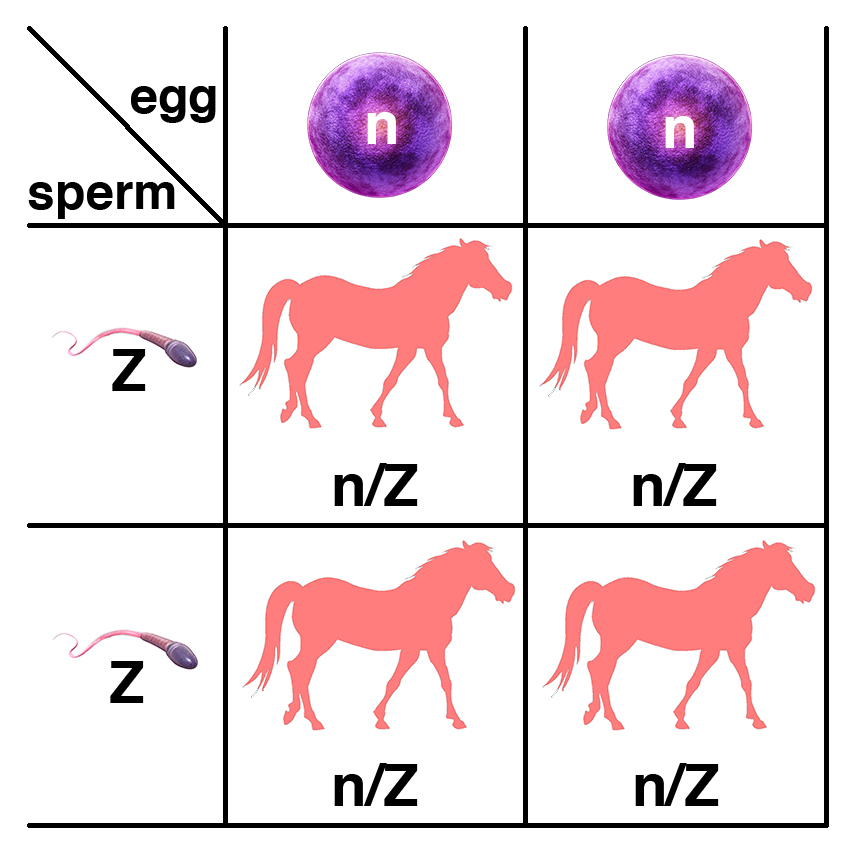
If a horse with two copies of the semidominant allele (Z/Z) is bred to a normal horse (n/n), all of the foals will have one copy of the semidominant allele (n/Z) and will be mildly affected.
Citations
Myofibrillar Myopathy (MFM)
Back to TopSummary
Myofibrillar Myopathy (MFM) is a form of exercise intolerance. The clinical signs manifesting during or after exercise resemble other types of exertional rhabdomyolysis. During an episode, horses are reluctant to move, experience pain, stiffness, and tremors, and sweat profusely. Serum creatine kinase (CK) and aspartate aminotransferase (AST) may be elevated, indicating muscle damage. In severe episodes, pigmenturia (coffee-colored urine due to the presence of myoglobin), inability to stand, and death can occur. These symptoms resemble other forms of exercise intolerance such as Polysaccharide Storage Myopathy (PSSM), Recurrent Exertional Rhabdomyolysis (RER), and Vacuolar Myopathy (VM).
Myofibrillar Myopathy (MFM) is distinct from these other forms of exercise intolerance with respect to findings on muscle biopsy. Histopathology shows ectopic accumulation of cytoskeletal proteins (e.g. desmin) and Z-disc degeneration. Because this finding is only made when anti-desmin staining is used, muscle biopsies on the same horses performed without desmin staining would likely be scored as PSSM2. As in PSSM2, focal myofibrillar disruption with accumulation of glycogen particles is seen. The condition has been observed in a subset of Arabian horses with exercise intolerance. Initial descriptions of MFM classified it as a subtype of Recurrent Exertional Rhabdomyolysis (RER) because it was a form of exercise intolerance observed in Arabians and Thoroughbreds. Researchers at EquiSeq currently think that it more closely resembles Polysaccharide Storage Myopathy, type 2 (PSSM2).
Researchers at EquiSeq have identified semidominant alleles of two genes that causes MFM in Thoroughbreds and related breeds. The results are not yet published in a peer-reviewed academic journal. Prior to publication, the variants have been termed P3 and P4. These are missense alleles of FLNC and MYOZ3, respectively.
The P3 and P4 variants are also found in horses diagnosed with PSSM2 by muscle biopsy when desmin staining is not used. The P2 variant of MYOT associated with PSSM2 has not yet been observed alone in horses diagnosed with MFM; both n/P3 and n/P4 horses that are free of other PSSM2 variants have been diagnosed with MFM.
Horses that carry one copy of either variant (n/P3 or n/P4) have a predisposition to develop MFM (also diagnosed as PSSM2). Horses with two copies of the variant (P3/P3 or P4/P4) are more strongly affected, with more severe symptoms and an earlier age of onset. Dietary therapy (a high protein diet) and a specific exercise regimen may help to manage the symptoms.
Date of Last Update: 03/28/2018
Results
Understanding the Results
Results of the genetic test for MFM are presented as shown below.
| Myofibrillar Myopathy | ||
|---|---|---|
| n/n | Clear | This horse tested negative for P3. The horse will not pass on the defect to its offspring. |
| n/P3 | Affected | Both the normal and mutant alleles are present. This horse is positive for the P3 variant and may develop symptoms of exercise intolerance. |
| P3/P3 | Affected | This horse carries two copies of the P3 variant. The horse is expected to develop symptoms of exercise intolerance. |
| Myofibrillar Myopathy | ||
|---|---|---|
| n/n | Clear | This horse tested negative for P4, the MFM-predisposing variant. The horse will not pass on the defect to its offspring. |
| n/P4 | Affected | Both the normal and mutant alleles are present. This horse is positive for the P4 variant and may develop symptoms of exercise intolerance. |
| P4/P4 | Affected | This horse carries two copies of the P4 variant. The horse is expected to develop symptoms of exercise intolerance. |
Horses carrying the P3 or P4 variants develop symptoms of exercise intolerance as they age. The age of onset and the severity of symptoms is variable.
What You Can Do
Horses that test positive for P3 or P4 should receive dietary supplementation with complete protein (whey), complementary protein (soy) or with specific amino acids that are typically limiting in plant protein (lysine, methionine, and threonine). This is a management strategy and not a treatment or cure.
Disease Name and Genes
MFM is associated with semidominant genetic variants (P3 and P4) of FLNC and MYOZ3, respectively.
| Horse Genes Associated with MFM and Human Diseases Associated with Variants of the Orthologous Genes | Variant | Inheritance | Gene Symbol | Gene Name | Associated Human Disease |
|---|---|---|---|---|
| P3 | Semidominant | FLNC | Filamin C | Myofibrillar Myopathy 5 |
| P3 | Semidominant | FLNC | Filamin C | Distal Myopathy 4 |
| P4 | Semidominant | MYOZ3 | Myozenin 3 | none |
Inheritance
MFM is associated with semidominant variants (P3 and P4) of FLNC and MYOZ3, respectively. The semidominant alleles are abbreviated as P3 and P4, with the recessive wild-type alleles abbreviated as n.
The illustrations below show the inheritance of P3 as an example. P3 and P4 are inherited in the same way. The two variants are inherited independently, and a horse may have more than one, so a horse may have the genotype n/P3 n/P4.
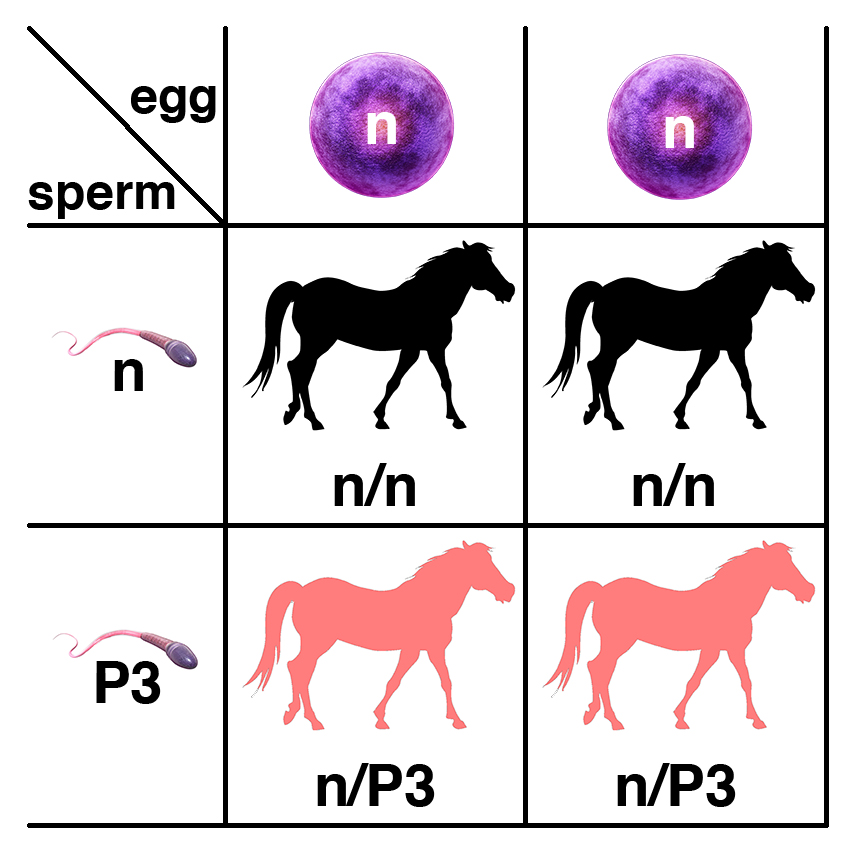
A horse with one copy of the semidominant allele (n/P3) will potentially have symptoms. If this horse is bred to a normal horse (n/n), each foal has a 50% chance of having one copy of the semidominant allele (n/P3) and a 50% chance of having two copies of the normal allele (n/n).

If two horses, each with one copy of the semidominant allele (n/P3), are bred, each foal has a 25% chance of having two copies of the normal allele (n/n), a 50% chance of having one copy of the semidominant allele (n/P3), and a 25% chance of having two copies of the semidominant allele (P3/P3). Because horses with one copy (n/P3) or two copies (P3/P3) of the semidominant allele are affected, each foal will have a 75% chance of potentially being affected.

If a horse with two copies of the semidominant allele (P3/P3) is bred to a normal horse (n/n), all of the foals will have one copy of the semidominant allele (n/P3) and will potentially be affected.
Citations
Myotonia
Back to TopSummary
Myotonia is a neuromuscular disorder that slows the relaxation of muscles after voluntary contraction or electrical stimulation. An inherited form of Myotonia was seen in a New Forest pony, which displayed symptoms including difficulty rising to its feet, abnormal vocalization, stiff gait, and rigidity of limbs, neck, and trunk. There is no effective treatment.
Date of Last Update: 08/02/2016
Results
Understanding the Results
Results of the genetic test for Myotonia are presented as shown below.
| Myotonia | ||
|---|---|---|
| N/N | Clear | This horse tested negative for Myotonia. |
| N/Myo | Carrier | Both the normal and mutant alleles are present. This horse is positive for the Myotonia mutation but will not develop symptoms. |
| Myo/Myo | Affected | This horse carries two copies of the Myotonia mutation and will develop the disease. |
Disease Name and Genes
Myotonia is caused by an A to C substitution in the skeletal muscle chloride channel gene CLCN1, resulting in a missense mutation in which an Aspartic Acid (D) codon is changed to an Alanine (A) codon at position 592 (CLCN1-D592A).
Inheritance
Myotonia resulting from the CLCN1-D592A variant shows a recessive mode of inheritance. The recessive allele is abbreviated here as Myo, with the dominant wild-type allele abbreviated as N.
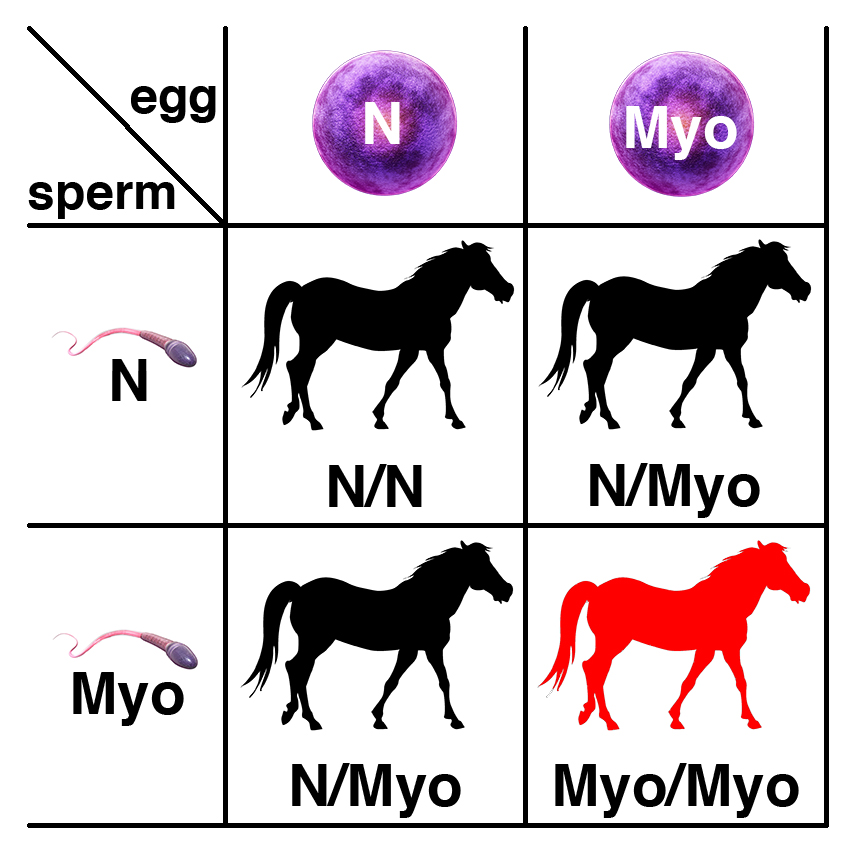
Carriers of the recessive allele (N/Myo) have no symptoms of the disease. If two carriers are bred, each foal has a 25% chance of having two copies of the normal allele (N/N), a 50% chance of being a carrier (N/Myo), and a 25% chance of being affected (Myo/Myo).

If a carrier of the recessive allele (N/Myo) is bred to a normal horse (N/N), each foal has a 50% chance of having two copies of the normal allele (N/N) and a 50% chance of being a carrier (N/Myo).
Citations
Naked Foal Syndrome (NFS)
Back to TopResults
Citations
Ovotesticular Disorder of Sexual Development
Back to TopSummary
Sex determination in mammals is dependent on sex chromosome composition, with XX animals being female and XY animals being male. In the absence of the Y-chromosome male determining gene SRY, no androgen is produced in the developing gonad, and the embryo develops as a female. In the presence of the Y-chromosome male determining gene SRY, the developing gonad produces androgen, and the embryo develops as a male. De novo deletions of the SRY gene in a mosaic gonad (the consequence of somatic mutation) will result in the transmission to chromosomally male (XY) offspring of Y chromosomes that fail to initiate male development as embryos. These will develop as sterile phenotypic females.
Sterile phenotypic females are obviously unsuitable for breeding, but are otherwise healthy.
Date of Last Update: 04/04/2016
Results
Understanding the Results
Phenotypic females that are XY by karyotyping or by the discovery of lack of heterozygosity for X-linked neutral variation outside of the pseudoautosomal region are potentially affected by Ovotesticular Disorder of Sexual Development (DSD). Absence of SRY by PCR or whole genome sequencing would confirm that the horse is 64, XY, SRY-negative.
Disease Name and Genes
Ovotesticular Disorder of Sexual Development (DSD) is a term applied to phenotypic sex reversal in 64, XY individuals that test negative for the Y-linked male determining gene SRY. Infertile phenotypic females with DSD that are 64, XY karyotypically lack the SRY gene as scored by attempted PCR amplification. The precise nature of the SRY mutation is unknown, but presumed to be a deletion.
Inheritance
Ovotesticular Disorder of Sexual Development (DSD) is transmitted as a de novo mutation from stallions to their sons, who appear as phenotypic females.

Stallions with a de novo mutation resulting in a Y chromosome that lacks the male-determine gene SRY have a mosaic germ line, with some of their sperm transmitting a normal Y chromosome (here shown as Y) and some of their sperm transmitting a Y chromosome lacking SRY (here shown as Y-SRY-). Some of the sons of these stallions will be normal colts (X/Y), while some will be 46, XY, SRY-negative phenotypic females (X/Y-SRY-) that are sterile. Note that in this diagram, the stallion produces two kinds of sperm carrying the Y chromosome, either a normal Y chromosome (Y), or a Y chromosome bearing a deletion of the SRY gene (Y-SRY-). Germ line mosaicism of this type is extremely rare, and most stallions produce only sperm bearing a normal Y chromosome.
Citations
Polysaccharide Storage Myopathy (PSSM)
Back to TopSummary
Polysaccharide Storage Myopathy (PSSM) is a form of exercise intolerance. The clinical signs manifesting during or after exercise resemble other types of exertional rhabdomyolysis. During an episode, horses are reluctant to move, experience pain, stiffness, and tremors, and sweat profusely. Serum creatine kinase (CK) and aspartate aminotransferase (AST) are elevated, indicating muscle damage. In severe episodes, pigmenturia (coffee-colored urine due to the presence of myoglobin), inability to stand, and death can occur. These symptoms resemble other forms of exercise intolerance such as Recurrent Exertional Rhabdomyolysis (RER), Myofibrillar Myopathy (MFM), and Vacuolar Myopathy (VM). Dietary therapy (elimination of sugars and the addition of protein and fats) and a specific exercise regimen can help to manage symptoms.
Date of Last Update: 08/01/2016
Results
Citations
McCue ME et al. (2008). "Glycogen synthase (GYS1) mutation causes a novel skeletal muscle glycogenosis." Genomics. 91(5):458-66. PMID: 18358695.Racing performance
Back to TopResults
Citations
Racing performance, sprint
Back to TopResults
Citations
Recurrent Exertional Rhabdomyolysis (RER)
Back to TopSummary
Recurrent Exertional Rhabdomyolysis (RER) is a form of exercise intolerance. The clinical signs manifesting during or after exercise resemble other types of exertional rhabdomyolysis. During an episode, horses are reluctant to move, experience pain, stiffness, and tremors, and sweat profusely. Serum creatine kinase (CK) and aspartate aminotransferase (AST) are elevated, indicating muscle damage. In severe episodes, pigmenturia (coffee-colored urine due to the presence of myoglobin), inability to stand, and death can occur. These symptoms resemble other forms of exercise intolerance such as Polysaccharide Storage Myopathy (PSSM), Myofibrillar Myopathy (MFM), and Vacuolar Myopathy (VM). Dietary therapy (elimination of sugars and the addition of protein and fats) and a specific exercise regimen can help to manage symptoms.
Episodes of RER are characterized by very high levels of serum CK and AST (values over 10,000, where the baseline value is under 500) and by myoglobinuria (coffee-colored urine), shown in the image below.

RER is distinguished from other forms of exercise intolerance by a specific defect that is evident using a research technique that is not available as a routine diagnostic method. Living muscle tissue from affected horses is more sensitive to caffeine- and halothane-induced contracture than is muscle tissue from unaffected horses. Horses with RER have an alteration in muscle cell calcium regulation.
Horses with RER exhibit episodes of tying-up that are induced by stress. Stress can include transport and other changes in routine. Horses with RER are often described as having an excitable temperament.
Date of Last Update: 05/29/2018
Results
Understanding the Results
Researchers at EquiSeq have identified Px, a genetic variant of CACNA2D3, that appears to be associated with RER. A hair test for this variant is available as an investigational test.
Results of the genetic test for RER are presented as shown below.
| Recurrent Exertional Rhabdomyolysis (RER) | ||
|---|---|---|
| n/n | Clear | This horse tested negative for Px. The horse will not pass on the defect to its offspring. |
| n/Px | Affected | Both the normal and mutant alleles are present. This horse is positive for the Px mutation and may develop symptoms of RER. |
| Px/Px | Affected | This horse carries two copies of the Px mutation and may develop symptoms of RER. |
Disease Name and Genes
Recurrent Exertional Rhabdomyolysis (RER), as seen in Arabians and Thoroughbreds, appears to be inherited, although the phenotype has not yet been assigned to a gene in the published literature. Affected sires have been shown to produce both affected and unaffected offspring, including colts, supporting an autosomal dominant mode of inheritance. Genome-wide association studies (GWAS) have identified several genomic regions that are associated with RER, although candidate genes have not been identified in the published literature. The genes RYR1, CACNA1S, and ATP2A1 have been excluded as candidate genes.
Researchers at EquiSeq have identified Px, a semidominant genetic variant of CACNA2D3 that appears to be associated with RER.
The Px mutation is a synonymous substitution in CACNA2D3, a gene that encodes a molecular chaperone required for the assembly of the voltage-gated calcium channel, the voltage sensor of muscle. The synonymous substitution caused by the Px mutation is near a weak exonic splicing enhancer, suggesting that the Px mutation may affect pre-mRNA processing of the CACNA2D3 primary transcript.
The Px mutation is in linkage disequilibrium with the risk alleles of three single-nucleotide polymorphisms (SNPs) that are associated with RER in some related Thoroughbreds but not others in the published literature. This suggests that Px requires another genetic component, which has not yet been identified.
A hair test for the Px variant is available as an investigational test. The semidominant allele is abbreviated as Px, with the wild-type allele abbreviated as n.

A horse with one copy of the dominant allele (n/Px) will potentially have symptoms. If this horse is bred to a normal horse (n/n), each foal has a 50% chance of having one copy of the semidominant allele (n/Px) and a 50% chance of having two copies of the normal allele (n/n).

If two horses, each with one copy of the dominant allele (n/Px), are bred, each foal has a 25% chance of having two copies of the normal allele (n/n), a 50% chance of having one copy of the semidominant allele (n/Px), and a 25% chance of having two copies of the semidominant allele (Px/Px). Because horses with one copy (n/Px) or two copies (Px/Px) of the dominant allele are affected, each foal will have a 75% chance of being affected.
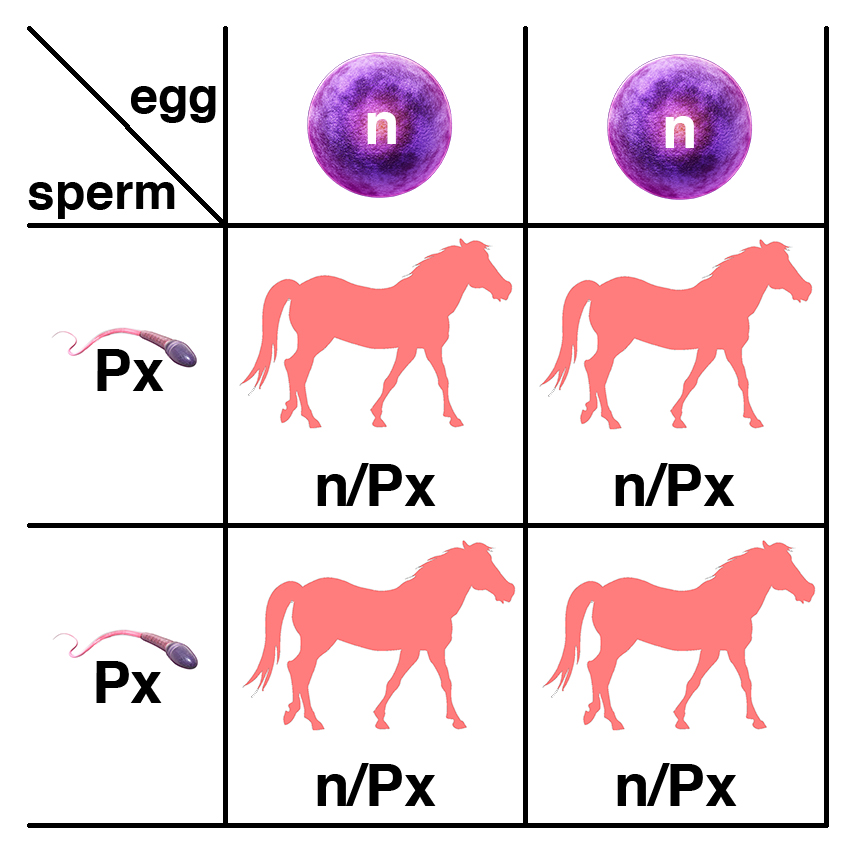
If a horse with two copies of the semidominant allele (Px/Px) is bred to a normal horse (n/n), all of the foals will have one copy of the semidominant allele (n/Px) and will be affected.
Inheritance
Researchers at EquiSeq have identified Px, a semidominant genetic variant of CACNA2D3 that appears to be associated with RER. The semidominant allele is abbreviated as Px, with the wild-type allele abbreviated as n.
Citations
Severe Combined Immune Deficiency (SCID), Autosomal
Back to TopSummary
Severe Combined Immune Deficiency (SCID) is an inherited defect of the immune system seen in Arabians. Horses with this condition are susceptible to infections shortly after birth, and typically die within six months. There is no effective treatment.
Date of Last Update: 08/02/2016
Results
Understanding the Results
Results of the genetic test for Severe Combined Immune Deficiency (SCID) are presented as shown below.
| Severe Combined Immune Deficiency (SCID) | ||
|---|---|---|
| N/N | Clear | This horse tested negative for SCID. |
| N/SCID | Carrier | Both the normal and mutant alleles are present. This horse is positive for the SCID mutation but will not develop symptoms. |
| SCID/SCID | Affected | This horse carries two copies of the SCID mutation and will die of the disease. |
Disease Name and Genes
Severe Combined Immune Deficiency (SCID) results from a five base deletion in the gene encoding the catalytic subunit of the DNA-dependent protein kinase (DNA-PKcs). The deletion causes a shift in the reading frame; three incorrect amino acids occur before a termination codon is reached at codon 3160. The frameshift mutation causes a 967 amino acid truncation, which deletes the binding domain for the Phosphoinositide 3-kinase (PI3K) protein. This results in a severe defect in V(D)J rearrangement, the mechanism used to form the coding region of Ig and TCR variable regions in the generation of antibody diversity.
Inheritance
Severe Combined Immune Deficiency (SCID) caused by a mutation in the DNA-PKcs gene is inherited as a recessive trait. The recessive allele is commonly abbreviated as SCID, with the dominant wild-type allele abbreviated as N.
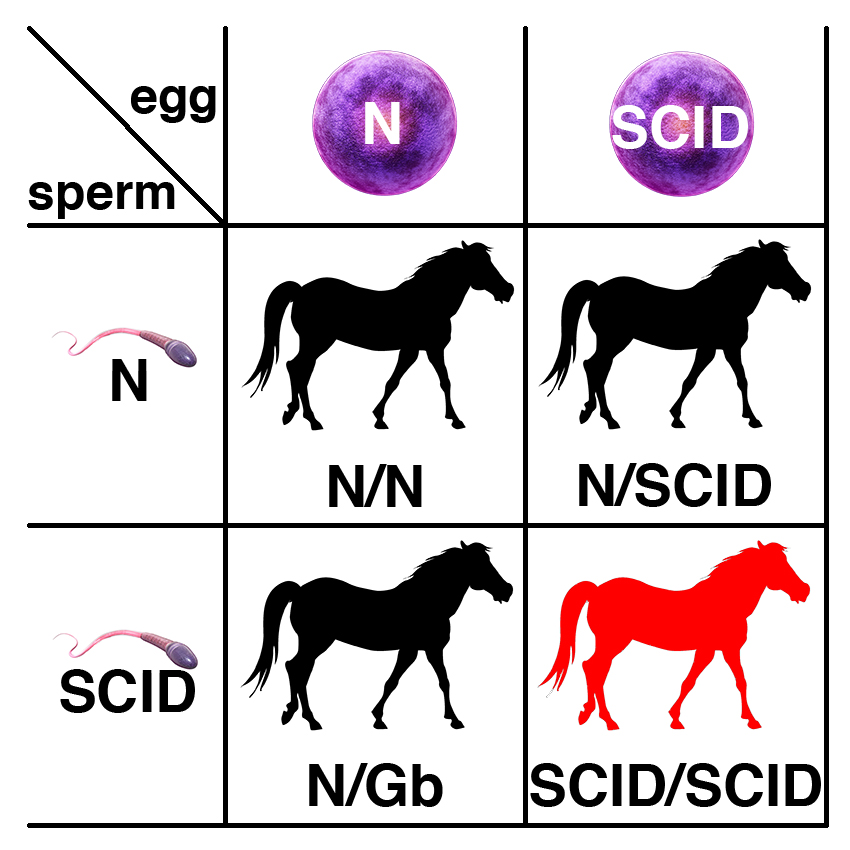
Carriers of the recessive allele (N/SCID) have no symptoms of the disease. If two carriers are bred, each foal has a 25% chance of having two copies of the normal allele (N/N), a 50% chance of being a carrier (N/SCID), and a 25% chance of being affected (SCID/SCID).
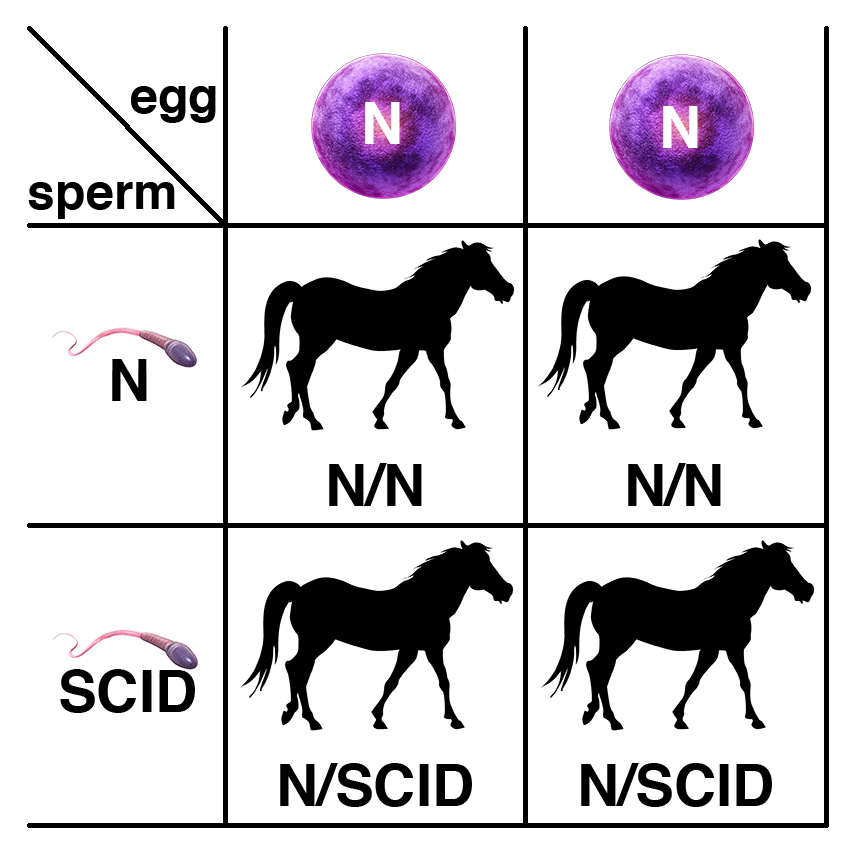
If a carrier of the recessive allele (N/SCID) is bred to a normal horse (N/N), each foal has a 50% chance of having two copies of the normal allele (N/N) and a 50% chance of being a carrier (N/SCID).
Citations
Show jumping
Back to TopResults
Citations
Splashed White Overo (SWO)
Back to TopSummary
Splashed White Overo (SWO) is term applied to mutations in different genes that are dominant modifiers of coat color, producing white patterning. The variants responsible for two forms of Splashed White Overo are recessive embryonic lethals. Embryos homozygous for either of these mutations cannot complete development.
Splashed White 2 (SW2) results from a mutation in PAX3 (PAX3-C70Y) and is seen in Quarter Horses. Splashed White 3 (SW3) results from a mutation in MITF (MITF-C280Sfs*20), and is seen in Quarter Horses.
Date of Last Update: 04/04/2016
Results
Citations
Thrombasthenia
Back to TopSummary
Glanzman Thrombasthenia is an inherited bleeding disorder marked by a failure of platelet aggregation. Affected horses bleed excessively and show a lack of clotting. There is no effective treatment.
Date of Last Update: 08/02/2016
Results
Understanding the Results
Results of the genetic test for Glanzman Thrombasthenia are presented as shown below.
| Glanzman Thrombasthenia (GT) | ||
|---|---|---|
| N/N | Clear | This horse tested negative for GT. |
| N/GT | Carrier | Both the normal and mutant alleles are present. This horse is positive for the GT mutation but will not develop symptoms. |
| GT/GT | Affected | This horse carries two copies of the GT mutation and will develop the disease. |
Disease Name and Genes
Two alleles of the gene encoding platelet glycoprotein IIb, Integrin, Alpha-2B (ITGA2B), are associated with Glanzman Thrombasthenia.
Glanzman Thrombasthenia results from a G to C substitution in exon 2 of ITGA2B that causes the replacement of an Arginine (R) with a Proline (P) at codon 72 (ITGA2B-R72P).
Glanzman Thrombasthenia also results from a 10 base-pair deletion that spans the splice site between exon 11 and intron 11 identified in a Quarter Horse. This mutation is predicted to result in a lack of splicing of intron 11 and inclusion of a termination codon 50 bp downstream of the deletion. Lack of expression of this mRNA is most likely the result of nonsense-mediated decay.
Inheritance
Glanzman Thrombasthenia resulting from mutations in the ITGA2B gene is inherited as a recessive trait. The recessive allele is here abbreviated as GT, with the dominant wild-type allele abbreviated as N.

Carriers of the recessive allele (N/GT) have no symptoms of the disease. If two carriers are bred, each foal has a 25% chance of having two copies of the normal allele (N/N), a 50% chance of being a carrier (N/GT), and a 25% chance of being affected (GT/GT).

If a carrier of the recessive allele (N/GT) is bred to a normal horse (N/N), each foal has a 50% chance of having two copies of the normal allele (N/N) and a 50% chance of being a carrier (N/GT).
Citations
Vacuolar Myopathy (VM)
Back to TopSummary
Vacuolar Myopathy (VM) is a very rare form of exercise intolerance, with only one case reported in the published literature. The clinical signs manifesting during or after exercise resemble other types of exertional rhabdomyolysis. During an episode, horses are reluctant to move, experience pain, stiffness, and tremors, and sweat profusely. Serum creatine kinase (CK) and aspartate aminotransferase (AST) are elevated, indicating muscle damage. In severe episodes, pigmenturia (coffee-colored urine due to the presence of myoglobin), inability to stand, and death can occur. These symptoms resemble other forms of exercise intolerance such as Polysaccharide Storage Myopathy (PSSM), Recurrent Exertional Rhabdomyolysis (RER), and Myofibrillar Myopathy (MFM).
Vacuolar Myopathy (VM) is distinct from these other forms of exercise intolerance with respect to findings on muscle biopsy. Histopathology shows abundant sarcoplasmic vacuoles of various sizes containing proteinaceous debris. Many vacuoles are devoid of glycogen and there is no amylase-resistant polysaccharide. Vacuoles do not stain for mitochondrial succinate dehydrogenase, mitochondrial cytochrome oxidase, or acid phosphatase.
Vacuolar Myopathy (VM) is presumed to be an inherited condition, although the genetic basis is currently unknown.
Date of Last Update: 08/02/2016
Results
Inheritance
The mode of inheritance of Vacuolar Myopathy (VM) is currently unknown.
Citations
Warmblood Fragile Foal Syndrome (WFFS)
Back to TopSummary
Warmblood Fragile Foal Syndrome (WFFS) is an inherited condition causing thin, fragile skin. Affected foals may be born with skin lesions and an open abdomen and are typically euthanized at birth or within a few days of birth. There is no effective treatment. The condition resembles Ehlers-Danlos syndrome in humans, which has multiple genetic causes. The skin defects resemble those seen in the Hereditary Equine Regional Dermal Asthenia (HERDA), which has a later onset of symptoms. There are also reports of Ehlers-Danlos-like syndromes of unknown origin in a number of breeds.
Warmblood Fragile Foal Syndrome is reported only in Warmbloods. The incidence of carriers is approximately 10%.
Date of Last Update: 08/02/2016
Results
Understanding the Results
Results of the genetic test for Warmblood Fragile Foal Syndrome (WFFS) are presented as shown below.
| Warmblood Fragile Foal Syndrome (WFFS) | ||
|---|---|---|
| N/N | Clear | This horse tested negative for WFFS. |
| N/FFS | Carrier | Both the normal and mutant alleles are present. This horse is positive for the WFFS mutation but will not develop symptoms. |
| FFS/FFS | Affected | This horse carries two copies of the WFFS mutation and will die of the disease. |
Disease Name and Genes
Warmblood Fragile Foal Syndrome (WFFS) results from a G to A substitution that causes the change from Glycine (G) to Arginine (R) at position 678 in the procollagen-lysine, 2-oxoglutarate 5-dioxygenase 1 (PLOD) gene (PLOD-G678R).
Inheritance
Warmblood Fragile Foal Syndrome (WFFS) resulting from the PLOD-G678R variant is recessive. The recessive allele is commonly abbreviated as FFS, with the dominant wild-type allele abbreviated as N.

Carriers of the recessive allele (N/FFS) have no symptoms of the disease. If two carriers are bred, each foal has a 25% chance of having two copies of the normal allele (N/N), a 50% chance of being a carrier (N/FFS), and a 25% chance of being affected (FFS/FFS).
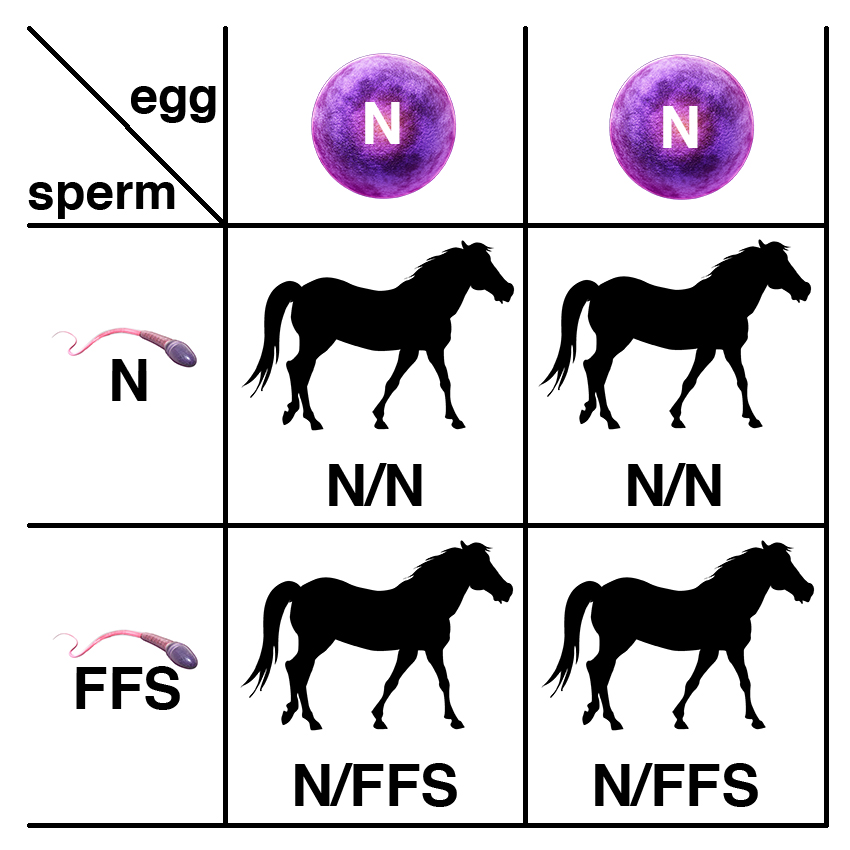
If a carrier of the recessive allele (N/FFS) is bred to a normal horse (N/N), each foal has a 50% chance of having two copies of the normal allele (N/N) and a 50% chance of being a carrier (N/FFS).





